Quality by Design in Generic Drug Development: Modern Science-Based Approaches
 Dec, 6 2025
Dec, 6 2025
What Quality by Design (QbD) Really Means for Generic Drugs
Quality by Design (QbD) isn’t just another buzzword in pharma. It’s a fundamental shift in how generic drugs are made - from guessing what works to knowing exactly why it works. Before QbD, companies would follow a fixed recipe: mix for 15 minutes, compress at 12 kN, dry at 45°C. If the final tablet passed lab tests, it was approved. No questions asked. But if it failed, the whole batch was thrown out. That’s reactive. QbD flips that. It asks: What makes this drug work? And then builds the process around that understanding.
The U.S. Food and Drug Administration (FDA) made QbD mandatory for all Abbreviated New Drug Applications (ANDAs) submitted after October 1, 2017. That’s not a suggestion. It’s a requirement. And it’s working. According to FDA data from 2022, generic drug approval rates jumped 23% since QbD became standard, and review times dropped by nearly five months per application. Why? Because QbD removes guesswork. It replaces trial-and-error with science.
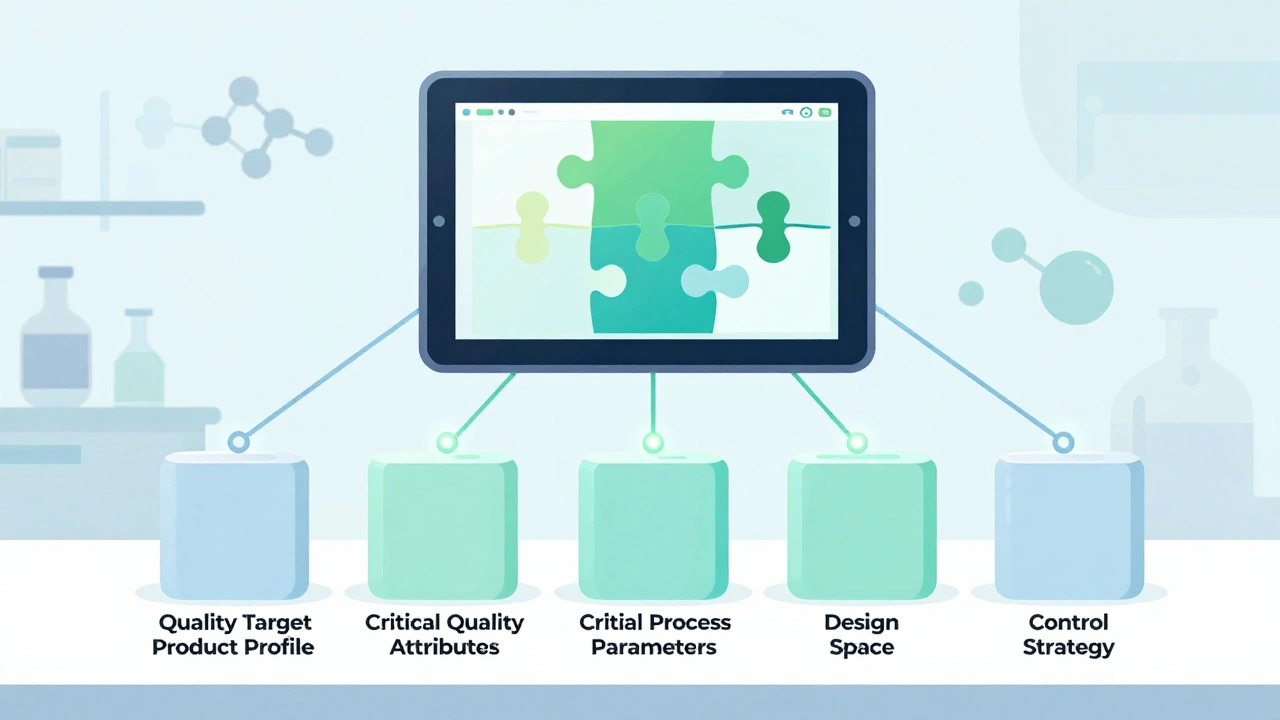
The Five Pillars of QbD in Generic Development
QbD isn’t one thing. It’s five connected parts, each built on data, not tradition.
- Quality Target Product Profile (QTPP) - This is the destination. What should the drug do? How fast should it dissolve? What impurities are acceptable? For generics, the FDA demands at least 95% similarity to the brand-name drug’s performance - especially in dissolution. If your tablet doesn’t release the medicine the same way as the original, it won’t be approved.
- Critical Quality Attributes (CQAs) - These are the measurable traits that define quality. For most generic tablets, that’s dissolution rate (f2 similarity >50), content uniformity (RSD ≤6.0%), and impurity levels (following ICH Q3B limits). You don’t test for these at the end. You design them in from the start.
- Critical Process Parameters (CPPs) - These are the levers you control during manufacturing. Granulation moisture? Compression force? Drying temperature? QbD doesn’t fix these at one number. It defines a working range. For example, compression force might be 10-15 kN. Outside that range, quality risks increase. Inside it? You’re safe.
- Design Space - This is where QbD gets powerful. It’s not a single point. It’s a multidimensional zone - a map of all the combinations of CPPs that guarantee quality. The FDA accepts design spaces based on data from over 100 simulated batches. Once approved, you can adjust parameters within this space without reapplying for permission. That’s flexibility. That’s savings.
- Control Strategy - How do you know you’re still in the design space? That’s where Process Analytical Technology (PAT) comes in. Near-infrared spectroscopy, Raman probes, real-time moisture sensors - these tools monitor the process as it happens. Eighty-seven percent of QbD users now use PAT. That cuts end-product testing by 35-60%. Less waste. Faster releases.
Why QbD Beats the Old Way
Traditional development is like driving with your eyes closed and only checking the speedometer every 10 miles. QbD is like having a GPS with live traffic updates.
Take scale-up. When you move from lab to factory, traditional methods often fail. A process that worked at 10 kg might crash at 1,000 kg. QbD-based processes are 28-42% more robust during scale-up, according to a 2023 Tufts study of 127 generic products. Why? Because you’ve mapped how each variable affects the outcome. You know what’s sensitive. You know what’s safe.
Regulatory headaches drop too. The FDA’s Office of Generic Drugs found QbD applications get 31% fewer Complete Response Letters (CRLs) - those dreaded “we need more data” letters. Approval timelines? 9.2 months for QbD vs. 13.9 months for traditional. That’s over four months faster. And when you get approved faster, you get to market sooner. Revenue starts flowing earlier.

The Hidden Costs and Real Challenges
QbD isn’t free. It’s not even cheap.
Initial development costs rise by 25-40%. Timelines stretch by 4-8 months. You need trained staff, advanced equipment, and software like MODDE Pro - which costs $15,000 per user per year. A single PAT system can set you back half a million dollars. For a simple immediate-release tablet, is that worth it? Sometimes, no.
Dr. James Polli from the University of Maryland warns against over-engineering. He’s seen companies spend $450,000 on complex DoE studies for a basic aspirin tablet - a product with a 50-year history and well-known behavior. That’s not innovation. That’s waste.
Complex generics are where QbD shines. Inhalers, transdermal patches, modified-release tablets - these are hard to copy. Traditional bioequivalence studies often fail to predict real-world performance. QbD fills that gap. But even here, 63% of failures come from poor understanding of how the formulation behaves in the body. If you can’t link your dissolution data to actual absorption, your design space is just a guess.
Real-World Wins and Hard Lessons
Companies that got it right are seeing real results.
Dr. Elena Rodriguez at Hikma Pharmaceuticals cut post-approval quality deviations from 14 per year to just 2 after implementing QbD for their generic esomeprazole. That saved $850,000 annually in investigations and recalls. Mylan (now Viatris) used their QbD control strategy for simvastatin to make 11 process changes during the pandemic without FDA approval - keeping supply flowing while others struggled.
But it’s not all smooth sailing. Dr. Mark Chen at Lupin said the first two QbD submissions nearly broke their team. Training took 120 person-hours per scientist. Documentation doubled. Morale dipped. The learning curve is steep. But after the first year, things stabilized. “We stopped fighting the system,” he said. “We started using it.”
The Generic Pharmaceutical Association found that 78% of companies saw better interactions with regulators after QbD. Fewer questions. Fewer delays. But 52% still struggle to justify design space boundaries for complex products. That’s where experience matters. You can’t just run a few DoE runs and call it done. You need deep understanding - and that takes time.
How to Start Getting QbD Right
You don’t need to rebuild your whole company overnight. Start smart.
- Use the Reference Listed Drug (RLD) - Reverse-engineer the brand-name product with advanced analytics. Characterize its dissolution, particle size, polymorph form. That cuts development time by 30%.
- Bracket your strengths - If you’re making 5mg, 10mg, and 20mg versions, you don’t need full studies for all three. Test the extremes. If those work, the middle is likely safe. That cuts studies by 45%.
- Think continuous manufacturing - Teva’s 2022 levothyroxine case showed a 28% boost in batch consistency using continuous production with QbD. It’s not just for big pharma anymore. Smaller players are adopting it too.
The FDA’s QbD Pilot Program has approved 87 submissions with a 92% first-cycle approval rate. That’s higher than the 78% for traditional apps. Free training modules are available. The Parenteral Drug Association offers certified courses with an 85% pass rate. Use them.
The Future: QbD Is Here to Stay
In 2018, only 38% of new generic applications included QbD. By 2022, that number was 74%. For complex generics? It’s 92%. The trend is clear.
The FDA’s new ICH Q14 guideline (effective December 2023) requires more robust analytical data - but rewards it with 40% faster validation. Their Emerging Technology Program has approved all 27 QbD-based continuous manufacturing applications submitted so far. By 2027, McKinsey predicts 95% of new generics will use QbD.
The WHO now includes QbD in its prequalification program. India’s top 10 generic makers invested $227 million in QbD capabilities in 2022. Even cost-sensitive markets are adapting - because if you want to sell globally, you need to meet global standards.
But don’t force it. For a $10-a-year generic with $2 million in revenue, spending $400,000 on QbD makes no sense. The Generic Pharmaceutical Association’s 2023 white paper says it best: QbD must be proportionate. Use it where it adds value. Skip it where it doesn’t. That’s not cutting corners. That’s smart science.
Final Thought: Quality Isn’t Tested - It’s Built
QbD doesn’t make generic drugs better because it adds more steps. It makes them better because it adds more understanding. You’re no longer copying a pill. You’re understanding how it works - and then building it to perform exactly as intended, every time.
The old way was about passing a test. The new way is about proving you know why it passes. And that’s what regulators, patients, and manufacturers all want: consistency. Confidence. Control.
Is QbD required for all generic drugs?
Yes, for all Abbreviated New Drug Applications (ANDAs) submitted to the FDA after October 1, 2017. The FDA made QbD a regulatory expectation, not an option. The European Medicines Agency (EMA) and Japan’s PMDA have similar requirements, especially for complex generics like inhalers and modified-release products.
Does QbD mean I need clinical trials for bioequivalence?
No. QbD replaces the need for clinical bioequivalence studies in most cases by using advanced in vitro testing. If your dissolution profile matches the reference drug (f2 >50), your impurity levels are within limits, and your design space is validated, regulators accept that as proof of equivalence. This saves time and money - and avoids exposing patients to unnecessary trials.
How long does QbD development take compared to traditional methods?
For a simple immediate-release tablet, QbD adds 4-8 months to the typical 18-24 month development cycle. For complex products like patches or inhalers, it can take 12-18 months just for the QbD phase. But the payoff is faster regulatory approval - QbD applications are approved in 9.2 months on average, compared to 13.9 months for traditional submissions.
Can small generic companies afford QbD?
It’s challenging, but possible. Start with high-value products - ones with higher margins or complex formulations. Use free FDA training resources. Partner with consultants who specialize in QbD for generics. Leverage RLD data to reduce testing. And focus on proportionate implementation - don’t over-invest in a $5 product. The goal isn’t perfection. It’s smart, science-based control.
What’s the biggest mistake companies make with QbD?
Treating QbD as a documentation exercise. Many companies run a few experiments, write a thick report, and think they’re done. But QbD isn’t about paperwork. It’s about understanding. If you don’t know why a change in moisture content affects dissolution, you haven’t done QbD. You’ve just added bureaucracy. Real QbD means you can predict outcomes, not just report them.
How does QbD help with supply chain disruptions?
QbD gives you flexibility. If a supplier changes, or a machine breaks down, you can adjust process parameters within your approved design space without waiting for FDA approval. One company made 11 changes during the pandemic without delays - keeping supply steady while others faced shortages. That’s resilience.
Kyle Oksten
December 8, 2025 AT 01:45Nancy Carlsen
December 9, 2025 AT 11:32Ted Rosenwasser
December 9, 2025 AT 12:06Helen Maples
December 10, 2025 AT 02:29Stacy here
December 11, 2025 AT 06:54Wesley Phillips
December 11, 2025 AT 13:17Olivia Hand
December 13, 2025 AT 09:40Desmond Khoo
December 14, 2025 AT 23:14Sam Mathew Cheriyan
December 15, 2025 AT 11:49Kyle Flores
December 16, 2025 AT 22:59Ryan Sullivan
December 18, 2025 AT 12:47Louis Llaine
December 20, 2025 AT 10:27Jane Quitain
December 20, 2025 AT 16:48Ashley Farmer
December 21, 2025 AT 20:44David Brooks
December 22, 2025 AT 09:27