Bioequivalence Studies: What the FDA Requires Generic Drug Manufacturers to Prove
 Jan, 23 2026
Jan, 23 2026
When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how does the FDA make sure that’s true? The answer lies in bioequivalence studies - the critical tests that prove a generic drug behaves the same way in your body as the original. These aren’t just paperwork. They’re real, controlled clinical trials that measure exactly how much of the drug enters your bloodstream and how fast it gets there.
Why Bioequivalence Matters More Than You Think
The FDA doesn’t require generic manufacturers to repeat the full clinical trials that brand-name companies ran. That’s because the active ingredient is the same. But here’s the catch: two pills can have identical ingredients and still behave differently in your body. One might dissolve too slowly. Another might be absorbed unevenly. That’s where bioequivalence comes in. The FDA’s definition is precise: bioequivalence means there’s no significant difference in the rate and extent that the active ingredient becomes available at the site of action. In plain terms - your body absorbs and uses the generic drug the same way it does the brand-name one. No more, no less. This isn’t theoretical. It’s backed by decades of science and real-world outcomes.The Two Rules: Pharmaceutical Equivalence and Bioequivalence
Before a generic drug even gets to the bioequivalence test, it must pass pharmaceutical equivalence. That means:- Same active ingredient
- Same strength
- Same dosage form (tablet, capsule, injection, etc.)
- Same route of administration (oral, topical, etc.)
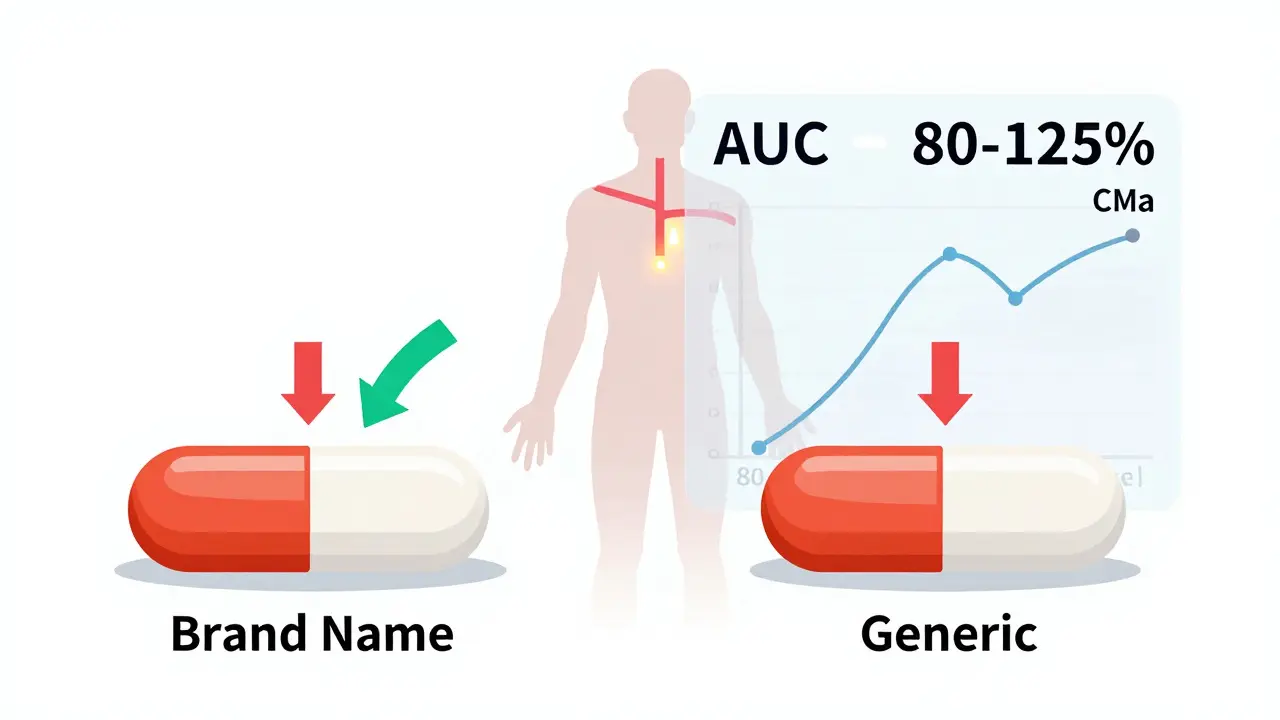
The 80/125 Rule: How the FDA Measures Bioequivalence
The gold standard for proving bioequivalence is a pharmacokinetic study in healthy volunteers. Typically, 24 to 36 people take both the generic and the brand-name drug on separate occasions, under fasting conditions. Blood samples are taken over time to track how the drug moves through the body. The FDA looks at two key numbers:- AUC - Area Under the Curve. This tells you how much of the drug your body absorbs overall.
- Cmax - Maximum Concentration. This shows how fast the drug reaches its peak level in your blood.
When the Rules Get Tighter: Narrow Therapeutic Index Drugs
Not all drugs are created equal. Some, like warfarin (a blood thinner) or levothyroxine (for thyroid disorders), have a very narrow window between an effective dose and a dangerous one. Even a small difference in absorption could be life-threatening. For these narrow therapeutic index drugs (NTIDs), the FDA uses stricter criteria. The 90% confidence interval must fall between 90% and 111%. That’s a much tighter range. Manufacturers of generics for these drugs face a higher bar - and for good reason. One wrong dose can lead to a stroke or a thyroid crisis.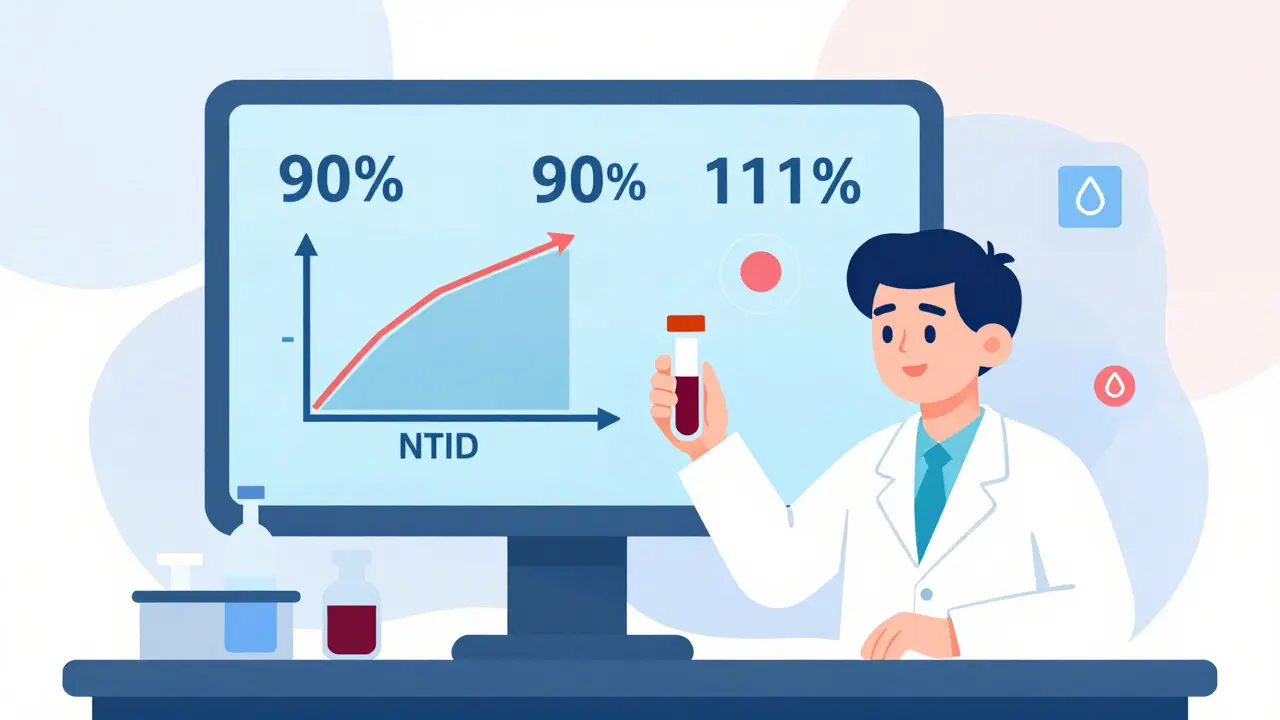
Biowaivers: When You Don’t Need a Human Study
Not every generic needs a full bioequivalence study. The FDA allows biowaivers - exemptions from human testing - for certain products where the risk of difference is extremely low. Examples include:- Oral solutions where the active ingredient is already dissolved and absorbed the same way as the brand
- Topical products meant to work on the skin, not in the bloodstream
- Ophthalmic or otic drops with identical ingredients and concentrations
- Q1: Same active and inactive ingredients
- Q2: Same dosage form and strength
- Q3: Same physicochemical properties (like pH, solubility, dissolution rate)
Complex Drugs Are a Different Challenge
Not all drugs are simple pills. Topical creams, inhalers, injectables, and drug-device combos (like auto-injectors) are harder to match. The same 80/125 rule doesn’t always apply. For these complex products, the FDA is moving toward advanced tools:- In vitro release testing (IVRT) - measuring how the drug comes out of the product under lab conditions
- In vitro permeation testing (IVPT) - tracking how the drug moves through skin or tissue
- Physiologically based pharmacokinetic (PBPK) modeling - computer simulations of how the drug behaves in the body
Why So Many Applications Get Rejected
It’s not just about getting the science right. Many applications fail because of sloppy paperwork or bad study design. The FDA’s 2022 report showed only 43% of ANDA applications were approved on the first try. Why? Common mistakes:- Too few volunteers in the study
- Improper sample handling or storage
- Using outdated or unvalidated lab methods
- Not following the product-specific guidance (PSG)

The Cost and Time Behind the Science
Running a bioequivalence study isn’t cheap. A single study can cost between $500,000 and $2 million. That’s why many manufacturers outsource to specialized contract research organizations. The entire ANDA process - from development to approval - used to take 36 months. Now, it’s down to 14-18 months on average. But bioequivalence data is still the biggest bottleneck. If the study fails, the whole application gets delayed. The FDA’s Domestic Generic Drug Manufacturing Pilot Program is helping. If a company uses U.S.-made active ingredients and conducts bioequivalence testing in the U.S., they get faster review. It’s one way the FDA is trying to bring more manufacturing back home.What This Means for Patients
You might not think about bioequivalence when you fill a prescription. But it’s the reason you can pay $4 for a generic instead of $400 for the brand. Without these studies, generics wouldn’t be trusted. And without trust, they wouldn’t be used. Today, 90% of all prescriptions in the U.S. are filled with generics. They save the healthcare system billions every year. But that system only works because the FDA holds manufacturers to a strict, science-backed standard.What’s Next for Bioequivalence?
The FDA isn’t standing still. Under its GDUFA III program (2023-2027), it’s committed to:- Issuing 1,800 new product-specific guidances
- Refining standards for complex generics
- Harmonizing requirements with international agencies like the EMA
Alexandra Enns
January 24, 2026 AT 14:20Let me tell you something - the FDA’s 80/125 rule is a joke. I’ve seen generics that made me feel like I was taking a different drug entirely. My cousin went from brand-name Adderall to a generic and started having panic attacks. No one talks about this. The FDA doesn’t test for *how you feel*, just how much hits your blood. That’s not science - that’s corporate babysitting.
Marie-Pier D.
January 25, 2026 AT 01:06Hey, I get where you're coming from, but I’ve been on generics for 8 years now - thyroid meds, blood pressure, you name it. No issues. I think the system works better than people give it credit for. 🙏 Also, generics saved me over $3k last year. That’s not nothing.
Himanshu Singh
January 26, 2026 AT 00:09Philosophically, this is beautiful. The body doesn’t care about brand names. It only cares about molecules. The FDA’s system is a quiet triumph of empirical logic over marketing. We live in an age where people trust influencers more than pharmacokinetics - and yet, here we are, trusting science without even realizing it. 🤔
Jamie Hooper
January 26, 2026 AT 00:39so like… the 80/125 thing? sounds like they just picked numbers outta their ass. i mean, why not 75/130? or 90/110? who even decided this? some guy in a lab coat with a calculator and a coffee stain?
Don Foster
January 27, 2026 AT 11:12Anyone who thinks bioequivalence is simple hasn’t read the actual FDA guidances. The Q1-Q2-Q3 criteria alone require PhD-level understanding of dissolution profiles and polymorph stability. And yet, people think generics are just ‘copy-paste’ pills. Pathetic. You don’t know what you don’t know.
siva lingam
January 28, 2026 AT 11:41So you pay $4 for a pill that’s ‘bioequivalent’ but still gives you a headache? Cool. I’ll stick with the $400 one. At least when it kills me, I know I paid for the premium experience. 😴
Chloe Hadland
January 28, 2026 AT 13:45Honestly I never thought about this until I started taking a generic for my anxiety. I was nervous at first but it worked just fine. It’s wild how much science goes into something so small. Just… thank you to the people who make sure this stuff is safe.
Amelia Williams
January 29, 2026 AT 11:41Okay but imagine if EVERYTHING worked like this? Like, what if your car parts had to be bioequivalent to the OEM ones? Or your shoes had to match the original brand’s sole flex pattern? We’d be stuck paying $200 for sneakers forever. Generics are the unsung heroes of modern life. Let’s celebrate them.
Viola Li
January 31, 2026 AT 03:09So let me get this straight - you’re okay with the government approving pills based on statistical ranges instead of exact matches? That’s not medicine, that’s gambling with people’s lives. And you call this progress? I’m not impressed.
Marlon Mentolaroc
January 31, 2026 AT 20:32Fun fact: 90% of generics are fine. The other 10%? They’re the ones that get recalled. And guess what? They’re almost always from overseas factories with zero oversight. The FDA’s system isn’t broken - it’s just underfunded. Fix the supply chain, not the science.
Gina Beard
February 1, 2026 AT 13:43There’s a difference between equivalence and identity. The body isn’t a test tube. It’s a living, breathing mess of hormones, gut flora, and inherited metabolism. We reduce it to numbers because it’s easier. But that doesn’t make it true.
Phil Maxwell
February 3, 2026 AT 12:27I’ve worked in a pharmacy for 12 years. People freak out over generics. Then they take them. Then they come back saying ‘wow, this is just as good.’ I’ve never seen anyone die from a generic. Ever. The fear is louder than the evidence.
asa MNG
February 4, 2026 AT 09:08bro the FDA is just a puppet for big pharma 😭 i mean come on. they let the same companies that make the brand name drugs test the generics. that’s like letting the fox inspect the chicken coop. and don’t even get me started on the ‘biowaivers’ - that’s just corporate laziness with a fancy name 🤡💸
Josh McEvoy
February 6, 2026 AT 00:21so like… if the generic works, why do people still pay for the brand? just because it’s got a pretty logo? 😏